O bolezni
Fenilketonurija (PKU) je genetska bolezen, povezana z presnovno motnjo aminokislin, predvsem fenilalanina. Zaužitje navadne beljakovinske hrane v otrokovo telo vodi do kopičenja strupenih presnovnih produktov v tkivih. Te spojine nepovratno vplivajo na živčni sistem in možgane otroka ter povzročajo progresivno demenco.
V nekaterih državah je bolezen poimenovana po odkritelju in se imenuje Völlingova bolezen. Drugo ime, fenilpiruvična oligofrenija, kaže na patogenezo bolezni.
Statistika širjenja dedne bolezni se od države do države razlikuje. Torej, v Rusiji bolezen najdemo pri enem dojenčku od 5-10 tisoč novorojenčkov (odvisno od regije države). Najvarnejša država glede tveganja za razvoj fenilketonurije je Finska (1: 100.000). Turčija je na prvem mestu po pojavu genetske bolezni, eden od 2600 novorojenčkov ima oslabljen metabolizem fenilalanina.
Med bolniki s fenilketonurijo so deklice dvakrat bolj verjetne kot dečki. Dedovanje poteka avtosomno recesivno, kar pomeni prisotnost mutirajočega gena v družinah obeh staršev. Zdravi nosilci genetske napake se morda ne zavedajo lastnih značilnosti in nimajo nobenih zunanjih manifestacij. Situacija se zaplete, ko se lastniki mutacije poročijo in bodo kmalu dobili otroke. Tveganje za dojenčka s fenilketonurijo je v tem primeru 25%. Polovica otrok, rojenih tej družini, bo ostala asimptomatski nosilec dedne napake.

Čeprav lahko bolezen privede do hudih, nepopravljivih motenj v otrokovem razvoju in znatno poslabša kakovost otrokovega življenja, se lahko izognemo neprijetnim posledicam. Fenilketonurija je ena redkih dednih bolezni, ki jo je mogoče učinkovito zdraviti. S pravočasno sprejetimi ukrepi otrok skoraj vedno raste zdrav.
Zgodovinska referenca
Prvič je na dedno naravo demence sumil norveški biokemik in psihiater Ivar Asbjörn Fölling. Ob opazovanju skupine duševno zaostalih otrok je zdravnik opazil skupno klinično sliko in ugotovil spremembo kemične sestave urina bolnih bratov in sester. Če je biološki tekočini dodal raztopino železovega klorida, je zdravnik razkril spremembo barve urina in videz olivno zelenega odtenka. Prisotnost sprememb v analizah pri bližnjih sorodnikih je povzročila sum na dedne vzroke bolezni.
Ta metoda odkrivanja fenilketonurije v sodobnem svetu ni izgubila pomembnosti. Določanje fenilpiruvične kisline v urinu se imenuje Völlingov test v čast norveškega zdravnika.

Kako se bolezen razvije?
Brez dodatnih raziskav je nemogoče napovedati razvoj fenilketonurije pri novorojenčku. Otrok se ne razlikuje od svojih vrstnikov, izgleda kot popolnoma zdrav otrok. Do sprememb pride v telesu, ko se začne enteralno hranjenje, beljakovine v materinem mleku vstopijo v telo.
Vzrok za patologijo je pomanjkanje nekaterih encimov, ki sodelujejo pri pretvorbi esencialne aminokisline fenilalanina v tirozin. Tako se raven fenilalanina in njegovih derivatov v krvi in cerebrospinalni tekočini poveča. Presnovni proizvodi imajo toksičen učinek na možganske celice, motijo presnovo maščob in prenos živčnih impulzov med nevroni.
Hkrati se tirozin, potreben za normalno delovanje, ne sintetizira v ustrezni količini. Ta aminokislina je bistvenega pomena in aktivno sodeluje pri tvorbi hormonov, nevrotransmiterjev, melaninskega pigmenta.
Obstaja več kliničnih oblik fenilketonurije, za katere je značilno pomanjkanje določenega encima v jetrih. V 98% se pojavi PKU tipa I, povezan z gensko napako v 12. kromosomu. Bolezen se kaže v pomanjkanju fenilalanin-4-hidroksilaze in se dobro odziva na zdravljenje. II in III vrste bolezni so izjemno redke bolezni in so značilne za hude manifestacije, neučinkovitost dietne terapije pri popravljanju presnove aminokislin.
Simptomi klasične oblike fenilketonurije
Pogoste manifestacije
Če v porodnišnici niso opravili presejanja novorojenčkov, bolezen ostaja neprepoznana in potrebni ukrepi zdravljenja niso bili organizirani, se bo PKU pokazal v prvem letu otrokovega življenja. Starši bodo prve znake odstopanj od norme našli pri otroku pri starosti 2-6 mesecev, ko prej zdrav malček postane letargičen in apatičen ali obratno pretirano razdražljiv. Prehranjevanje se pogosto konča z regurgitacijo, na otrokovi koži pa se pojavijo manifestacije alergijskega dermatitisa.
Značilen videz
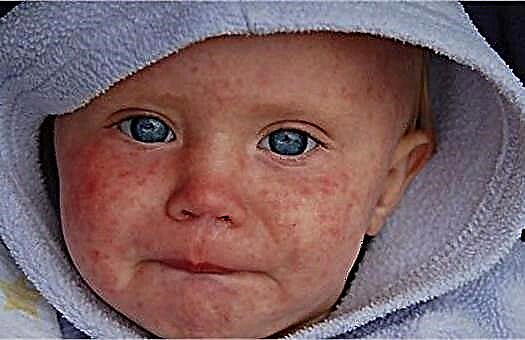
Ker kršitev presnove aminokislin vodi do zmanjšanja tvorbe melanina, v primeru razvoja bolezni koža, lasje in šarenica oči izgubijo pigment. Dojenčki z genetsko motnjo imajo svetle lase, tanko bledo kožo in modre oči. Značilen vonj znoja in urina drobtin lahko kaže na dedno bolezen. Mnogi zdravniki jo opisujejo kot "miško", povezana pa je s sproščanjem presnovnih produktov aminokislin z biološkimi tekočinami.

Poškodbe živčnega sistema
Prvi znaki sprememb v živčnem sistemu se kažejo v letargiji, zmanjšanju mišičnega tonusa drobtin. Pogosto se pojavi distonija, pojavijo se nehotene mišične kontrakcije, obsesivni gibi, krči. Možen je razvoj organskih patologij v možganih - mikrocefalija in hidrocefalus.
Skoraj 50% bolnikov s fenilketonurijo razvije epilepsijo. Včasih so značilni napadi prvi znak dedne bolezni. Paroksizme je težko zdraviti s konvencionalnimi antikonvulzivi in znatno zmanjšajo otrokovo kakovost življenja.
Razvojni zaostanek
S potekom bolezni dojenček hitro izgubi zanimanje za svet okoli sebe, postane apatičen. Trpi tudi telesni razvoj otroka - otrok pozno obvlada potrebne veščine. Zamuda v duševnem razvoju se pokaže, ko se pojavijo težave pri oblikovanju govora, zaostanek v duševnem razvoju.

Študije so pokazale, da se po letu napredovanja bolezni razvijejo velike spremembe v intelektualni dejavnosti. Drobtina nepovratno izgubi do 50 točk IQ letno, kar posledično povzroči razvoj oslabelosti, imbecilije ali idiotizma. Zato je čas začetka zdravljenja fenilketonurije zelo pomemben.
Nevrološke težave pri starejših otrocih se izražajo v hiperaktivnosti, pojavu stereotipov, obsesivnih gibih rok, obraza, jezika, zibanju. Včasih se duševne motnje kažejo v shizofrenih stanjih.
Huda razvojna zakasnitev vodi do pomanjkanja spretnosti pri hoji pri 30% bolnih otrok in alalije, nerazvitosti govora pri 60% bolnikov.
Vrste patologije
Prehodna oblika hiperfenilalaninemije
Bolezen ne poteka vedno klasično, včasih je povišanje ravni fenilalanina začasno in bolezen sama se ne razvije. Kratek porast koncentracije aminokislin v otrokovi krvi je lahko povezan z nezrelostjo otrokovih encimskih sistemov, kar je najbolj značilno za globoko prezgodaj rojene otroke. Takšni dojenčki zahtevajo podrobnejši pregled, da se ugotovi vzrok bolezni.
Pterin odvisne variante fenilketonurije
Približno 2% dojenčkov, ki so jim v porodnišnici diagnosticirali fenilketonurijo in jim je predpisana ustrezna prehrana, razvije nevrološke težave. Ti simptomi lahko kažejo na razvoj redke, od pterina odvisne oblike PKU (tip II, III po klasifikaciji) pri dojenčku. Klinične manifestacije atipičnih oblik PKU so podobne poteku klasične oblike, vendar simptomi napredujejo hitreje in dietna terapija nima želenega učinka.

Ogromno vlogo pri razvoju od pterina odvisnih oblik igra pomanjkanje nevrotransmiterjev, ki se pojavi zaradi kršitve presnove aminokislin. Te snovi so potrebne za prenos živčnih impulzov med živčnimi celicami, brez katerih normalno delovanje telesa ni mogoče.
Fenilketonurija II velja za bolj maligno od tipa III. Bruto presnovne motnje aminokislin vodijo do velike smrti živčnih celic. Dojenčki s progresivnim PKU tipa II pogosto umrejo v starosti od 2 do 3 let.
Materinska fenilketonurija
Otroci s PKU se ne rodijo vedno zdravim staršem. Če nosečnice z dednimi motnjami ne upoštevajo diete, obstaja velika verjetnost, da bodo rodile otroka s sindromom fenilketonurije pri materi. Strupene snovi prodirajo skozi posteljico od materinega telesa do ploda in motijo tvorbo otrokovih organov in sistemov. Otroci se rodijo z več prirojenimi malformacijami, mikrocefalijo, duševno in telesno zaostalostjo.
Diagnostika
Družinsko medicinsko genetsko svetovanje
Bodoči starši morajo biti še posebej previdni, če so v družinah katerega od njih obstajali primeri rojstva otrok s fenilketonurijo. Nemogoče je izključiti možnost rojstva otroka s PKU, tudi če se je bolezen pokazala pri daljnem sorodniku. Zahrbtnost bolezni je v asimptomatskem prevozu poškodovanega gena s strani popolnoma zdravih ljudi.
Vsi pari, ki v družini sumijo na dedne bolezni, se morajo že med načrtovanjem otroka posvetovati z genetikom. V Medicinsko genetskem centru (MGC) je s sodobnimi diagnostičnimi metodami mogoče identificirati prevoz mutantnega gena in izračunati tveganje za otroka s PKU in drugimi genetskimi boleznimi.
Invazivne diagnostične metode (horionska biopsija, amniocenteza, kordocenteza) lahko pomagajo ugotoviti bolezen še pred rojstvom otroka. V tem primeru se preuči genetski material, pridobljen iz ploda. Te metode so travmatične in lahko vodijo do spontanega splava. Zato je njihova uporaba upravičena le z dokazanim prenosom mutiranih genov pri starših in visokim tveganjem za PKU pri dojenčku.
Presejanje novorojenčkov

Pred odpustom iz porodnišnice novorojenčke množično pregledajo zaradi dednih bolezni. Za to pomembno študijo zdravstveni delavec odvzame kri iz otrokove pete in na filtrirni del preskusne površine nanese kapljice biološke tekočine. Vsaka kapljica krvi je namenjena odkrivanju ene od petih bolezni: fenilketonurija, cistična fibroza, prirojena hipotiroidizem, adrenogenitalni sindrom, galaktozemija.
Presejanje novorojenčkov je zelo pomemben in nujen pregled za vse otroke brez izjeme. Poteka popolnoma brezplačno in je namenjen ohranjanju zdravja naroda. Z zavrnitvijo manipulacije starši naredijo veliko napako, saj simptomi dednih bolezni pri novorojenčku niso vedno opazni. Živahne klinične manifestacije se pogosto pojavijo, ko je otroku že težko pomagati in so spremembe v telesu drobtin postale nepopravljive.
Ta metoda je zanesljiva pri odkrivanju fenilketonurije, če otrok prejme zadostno količino enteralne prehrane, materino mleko. Zato novorojenčke, ki so na oddelku za intenzivno nego, pozneje analizirajo. Čas vzorčenja se razlikuje tudi pri nedonošenčkih in nedonošenčkih. Za dojenčke, ki so se pojavili pravočasno, se priporoča komplet kapilarne krvi na 4. dan življenja. Študija o prezgodaj rojenih drobtinah je prestavljena na 7 dni.
Analizo je treba opraviti na tešče, kar bo zagotovilo natančnejši rezultat in povečalo diagnostično vrednost testa. Obrazec z otrokovimi kapljicami krvi in podatki o potnih listih staršev se pošljejo v laboratorij medicinsko-genskega svetovanja, kjer se opravi biokemijska študija.
Starši morajo biti pozorni na pravilnost izpolnjevanja obrazca, pravilnost svojih kontaktnih podatkov. Študija v povprečju traja 10 dni, družina pa je takrat navadno doma. Če se ugotovi pozitiven rezultat testa, bodo morali starši čim prej stopiti v stik z genskim centrom, nepravilni kontaktni podatki pa bodo odložili nadaljnji pregled otroka.
Pregled otroka v Moskvi
Za potrditev bolezni pri dojenčku in ugotavljanje njenega vzroka bo moral otrok opraviti večstopenjski postopek pregleda. Ponovljeni biokemični testi bodo pomagali potrditi zvišanje ravni aminokislin v krvi in sprejeti odločitev o potrebi po dieti. Izvaja se študija ravni fenilalanina in tirozina, aktivnosti jetrnih encimov, določanje presnovnih produktov aminokislin v urinu.
Nato se izvede molekularno genetska diagnostika, ki omogoča identifikacijo mutacije v genu RAS, odgovornega za razvoj fenilketonurije. S pomočjo teh metod je mogoče določiti asimptomatski prenos bolezni.
PKU zdravljenje
Najučinkovitejša metoda zdravljenja klasične oblike fenilketonurije se šteje za pravilno organizirano dietno terapijo. Le z izključitvijo živil, ki vsebujejo veliko količino aminokislin, iz prehrane drobtin lahko pričakujemo pozitivne rezultate zdravljenja.
Značilnosti dietne terapije
Pravila za izbiro živilskih izdelkov
Fenilketonurija pri otrocih je težka bolezen in le zdravnik lahko izbere pravo prehrano za dojenčka. Pri izbiri dovoljenih izdelkov in izračunu njihove količine zdravnik upošteva nekatere posamezne značilnosti:
- kakšna oblika bolezni je prisotna pri dojenčku;
- resnost bolezni, raven fenilalanina v krvi;
- starost drobtin;
- značilnosti fizičnega razvoja fidgetov;
- fiziološke potrebe rastočega organizma po beljakovinah;
- kako dojenček prenaša vnos majhne količine fenilalanina iz hrane.
Aminokislina fenilalanin je nenadomestljiva, kar pomeni, da jo lahko zaužijemo samo z beljakovinsko hrano. Odraščajoč otrok potrebuje zadostno količino hranil, saj približno 40% fenijalanina v prehrani porabi za izgradnjo lastnih beljakovin. Izkazalo se je, da je tudi nemogoče popolnoma izključiti nevarne izdelke iz prehrane drobtin. Zato se v prvih dneh dietne terapije v laboratoriju oceni, kako se telo drobtin odzove na majhen vnos aminokislin s hrano.
Ali je dojenje možno s fenilketonurijo?
Ker v večini primerov bolezen določimo v prvih tednih otrokovega življenja, se postavlja vprašanje o varnosti hranjenja otroka z materinim mlekom. Pravzaprav je dojenje nujno za normalen razvoj otroka, oblikovanje njegovega imunskega sistema. Toda njegova uporaba je upravičena le v primerih, ko se mati drži jasnih pravil in strogo nadzoruje količino hrane, ki jo poje dojenček.
Za varno hranjenje je bolje uporabiti sveže iztisnjeno materino mleko, katerega dovoljeno količino izračuna zdravnik na podlagi otrokove telesne teže in njegove potrebe po fenilalaninu.
Mlajši kot je otrok, večja je njegova potreba po aminokislini. Na primer, dovoljena količina fenilalanina za novorojenčka je 90 - 60 mg / kg telesne teže na dan, medtem ko se potrebna količina aminokisline za šolarja zmanjša na 10 - 20 mg / kg telesne teže. Količino fenilalanina v živilih lahko izračunamo glede na to, da 1 g beljakovin vsebuje približno 50 mg aminokisline.
Specializirane mešanice
Obstajajo posebej razviti izdelki za racionalno prehrano otrok s fenilketonurijo. Izdelane so na osnovi beljakovinskih hidrolizatov ali mešanice aminokislin, ki so v celoti ali delno brez fenilalanina. Prehrana z zdravilnimi mešanicami je namenjena polnjenju telesa s potrebnimi beljakovinami, zadostno zalogo hranil. Te formule se lahko uporabljajo kot nadomestki materinega mleka ali kot dodatek k glavni prehrani za starejše otroke.
Sestava medicinskih mešanic za otroke različnih starosti je različna. Na primer, za otroke, mlajše od enega leta, so primerni izdelki, kot so "Afenilak", "Lofenalak". Starejšim otrokom so prikazani "Tetrafen", "Brez fenila", "Maxamum - XP".

"Živilski semafor"
Otrok vsako leto raste in njegove potrebe po hrani se povečujejo. Čas je, da uvedete dopolnilna živila, poskusite nove jedi. Starši se morajo zavedati nevarnosti fenilalanina za otrokove možgane, izbrati samo hrano, ki jo odobri zdravnik.
Prehrana bolnega otroka se bistveno razlikuje od prehrane vrstnikov. Dopolnilno hranjenje se začne z vnosom zelenjave in sadja, nato je treba dodati testenine brez beljakovin, nekatere vrste žit. Zdravnik mora sestaviti otrokov prvi meni in nadzirati vsebnost aminokislin v krvi glede na spremembe v prehrani.
Vsi izdelki so razdeljeni v skupine glede na vsebnost fenilalanina v njih:
- rdeči seznam.
Na tem seznamu je priporočljivo popolnoma izločiti živila, saj so bogata z beljakovinami. To so meso in klavnični proizvodi, ribe, jajca, kruh, ajda, ječmenova žita, Herkulovi kosmiči;
- rumeni seznam.
Dovoljeno je uporabljati majhno količino izdelkov iz te skupine pod nadzorom krvnega testa. Morda uvedba koruznega in riževega drobljenca, krompirja, mlečnih izdelkov z nizko vsebnostjo beljakovin;
- zeleni seznam.
Ti izdelki so osnova za prehrano starejših otrok s fenilketonurijo: koruzna in riževa moka, večina zelenjave, sadja in jagodičja, sladkarije, maslo.
Ko uporabljate končne izdelke, morate natančno prebrati navodila na embalaži. Uživanje banan, suhega sadja in stročnic lahko poveča koncentracijo fenilalanina, zato je treba njihov vnos omejiti.

Druge terapije
Uporaba zdravil je upravičena le za zdravljenje pterin odvisnih oblik PKU, kadar prehrana nima želenega učinka. V takih primerih se uporabljata zdravili L-dopa in 5-hidroksitriptofan.
V drugih primerih je mogoče kot dodatek k prehrani uporabiti vitaminsko terapijo, mineralne komplekse. Po potrebi so predpisana nootropna zdravila, antikonvulzivi. Sodelovanje z logopedi, psihologi, masažo, fizioterapijo ima tudi pozitiven rezultat pri zdravljenju bolezni.
Napoved in preprečevanje
Potek klasične oblike bolezni bistveno ne vpliva na pričakovano življenjsko dobo osebe. Toda brez ustreznega zdravljenja hudi simptomi bolezni pomembno vplivajo na bolnikovo inteligenco in kakovost življenja. V primeru racionalne prehranske terapije in pravočasnega nadzora ravni fenilalanina v krvi se otroci s PKU ne razlikujejo od vrstnikov in se dobro prilagajajo v družbi. Atipične oblike bolezni pogosto vodijo v invalidnost in smrt otroka, saj zdravljenje redko doseže dober rezultat.
Strokovnjaki priporočajo upoštevanje diete skozi življenje človeka, vendar je možno nekaj olajšanja, ko otrok dopolni 18 let. Nosečnice s PKU morajo biti še posebej previdne, da se izognejo sindromu materine fenilketonurije pri otroku.
Preventiva zajema preprečevanje tesno povezanih zakonskih zvez, medicinsko in genetsko svetovanje družinam, ki jim grozi razvoj PKU, in množičen pregled novorojenčkov.

Zaključki
Otroška bolezen je velik stres in preizkus za mlade starše, še posebej, če je bolezen dedna. A fenilketonurija je ena redkih genskih bolezni, pri kateri posegi zdravljenja prinašajo dobre rezultate.
Starši morajo razumeti pomen presejanja novorojenčkov in zgodnje, dobro izbrane dietne terapije. Vredno je natančno upoštevati vsa priporočila lečečega zdravnika in potem tako grozljiva diagnoza, kot je fenilketonurija, ne bo postala stavek za otroka.